Austrian Society for Laboratory Medicine and Clinical Chemistry
| Labordiagnostik bei Coronavirus SARS-CoV-2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HOME | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DOWNLOAD Version 1.6, 07.11.2020 Significant changes compared to the previous version: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Due to the increasing number of COVID-19 cases in Austria, which are caused by the coronavirus SARS-CoV-2, the Austrian Society for Laboratory Medicine and Clinical Chemistry (ÖGLMKC) would like to summarize the cornerstones of laboratory diagnostics for COVID-19. The recommendations are also coordinated with the Austrian Society for Hygiene, Microbiology and Preventive Medicine (ÖGHMP). This summary corresponds to the current state of knowledge; new scientific knowledge of COVID-19 is currently being published according to sometimes greatly shortened peer review procedures and requires ongoing updating and a rational evaluation.
1. Diagnosis of a SARS-CoV-2 infection 1.1. Direct virus detection using PCR 1.2. Direct virus detection using an antigen test 1.3. Indirect virus detection using an antibody test 1.4. Definition of terms for the properties of diagnostic tests 1.1. Direct virus detection using PCR The laboratory diagnostic gold standard for the diagnosis of an infection with coronavirus SARS-CoV-2 is the direct virus detection from respiratory secretions by means of polymerase chain reaction (PCR) or other nucleic acid amplification techniques (NAT). 1.1.1. Sampling and transportationSamples are obtained from the upper respiratory tract using an oro or nasopharynx smear. It is particularly important to rinse the throat (with water) before taking an oropgharyngeal smear or to clear the nose by blowing the nose before taking a nasopharyngeal smear. This means that PCR inhibitors can be removed or reduced. Alternatively, samples from the deep respiratory tract (induced sputum, tracheal secretion or bronchoalveolar lavage) can also be used in the appropriate clinical situation. Coronavirus SARS-CoV-2 has also been detected by means of PCR in stool and in individual cases in urine, blood/plasma and liquor, but these materials are currently not recommended for the primary diagnosis of COVID-19 disease. Samples must be taken by appropriately trained personnel. The self-acceptance of samples is assessed critically by the ÖGLMKC. On the one hand, the quality of the sampling must be guaranteed to avoid false negative results. Alternative acceptance methods must therefore be fully validated according to medical standards. On the other hand, a clear identification of the patient is necessary, particularly if official decisions are to be derived from the test result. Coronaviruses are enveloped RNA viruses, the virus particles and their single-stranded RNA genome are therefore sensitive to surfactants and RNAses. For this reason, special requirements for sampling, storage and transport must be made in the preanalytics in order to avoid false-negative results. In the case of swabs, it must be ensured that suitable swabs and transport media are used for virus detection ("virus swabs" with appropriate transport medium or, if necessary, dry sterile swabs with a small amount (1 - 3 ml) of sterile NaCl solution suitable for molecular genetic analyzes, at least none Gel swab). The selection of a suitable swab should be checked in the course of establishing the test. After collection, the samples should be brought to the laboratory as soon as possible. If samples need to be stored, this can be done at 2-8 � C for a maximum of 3 days or according to the manufacturer's instructions. Samples must be sent as "biological substance, category B" of the UN no. 3373 according to the specifications of packing instruction P650. For longer transport times, the shipment should be refrigerated. 1.1.2. Sample handling in the laboratory for virus detectionThe handling of respiratory samples in the context of laboratory diagnostics of SARS-CoV-2 infections falls into the category "non-targeted activities" and should be restricted to specially trained laboratory personnel. Updated German-language guidelines for obtaining and handling potentially contagious sample material can be found on the websites of AGES (Austrian Agency for Health and Food Safety), the RKI (Robert Koch Institute) and ABAS (Committee for Biological Agents). In particular, we allow ourselves to refer to the recommendations for handling test material from Covid 19 positive / suspect patients in the laboratory of the Austrian Society for Hygiene, Microbiology and Preventive Medicine (ÖGHMP). LINK: https://www.oeghmp.at/media/empfipps_zum_umgang_mit_untersuchungsmaterial_von_covid-19-positiven-verdaechtigen_patienten_im_labor.pdfIn a nutshell, non-targeted activities, such as sample preparation and preparation or inactivation for molecular biological tests (PCR) are carried out under the conditions of biological safety level 2 (BSL-2). All activities that can lead to the release of droplets or aerosols with SARS-CoV-2, e.g. the opening of sample vessels with respiratory material must be carried out in a class 2 safety workbench. In addition to the general safety precautions such as protective gowns and gloves, breathing protection measures (at least FFP-2; filtering face piece, fine dust mask) are recommended to wear safety glasses. Targeted activities with SARS-CoV-2 (virus isolation, neutralization test or similar) may only be carried out by specially trained personnel in security level 3 (BSL-3) facilities. 1.1.3. PCR test1.1.3.1. General A number of commercial test systems from various suppliers are available for the PCR detection of SARS-CoV-2. Typically, viral nucleic acid (RNA) is extracted from the sample first. From this, after reverse transcription (RT), a PCR (or another NAT such as isothermal amplification) is carried out to detect virus-specific nucleic acids.
The availability of reagents for PCR tests and RNA extraction as well as smear systems has improved significantly. Both medical laboratories and manufacturers of commercial test systems continue to work on increasing the analysis capacities. The ÖGLMKC explicitly endorses and supports collaborations to further increase the analysis capacity, but points to the absolute necessity of uncompromising quality control of the SARS-CoV-2 PCR analysis in order to guarantee valid and reproducible test results for all patients. Successful participation in round-robin tests at regular intervals is an essential prerequisite for qualitative analysis. In Austria, the Austrian Society for Quality Assurance and Standardization of Medical Diagnostic Examinations (ÖQUASTA) offers round-robin tests for SARS-CoV-2 diagnostics. 1.1.3.2. Molecular genetic basisEstablished in-house and commercial test systems are mostly based on the detection of 2 gene sequences (targets) of SARS-CoV-2. One gene sequence is usually selective for the genus Betacoronavirus and one gene sequence is specific for the Cladus SARS-CoV-2. The test systems used to date usually detect 2 sequences of the following genes: N (nucleocapsid), E (envelope), S (spike) and RdRP (RNA-dependent RNA polymerase). According to the updated WHO Guidelines for Laboratory Analysis (03/20), if the spread of SARS-CoV-2 in a region is advanced, a simplified workflow with amplification of only one specific region (e.g. PCR detection of the E gene) can be used (https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/laboratory-guidance). In addition, an additional internal control that checks the nucleic acid extraction and amplification as well as the integrity of the reagents must ensure the validity of the test results. The described detection limit of the first PCR assays for SARS-CoV-2 is around 10 copies of viral nucleic acids per reaction. (J Clin Microbiol. 2020 Mar 4. pii: JCM.00310-20. Doi: 10.1128 / JCM.00310-20 .; Euro Surveill. 2020 Mar; 25 (9). Doi: 10.2807 / 1560-7917.ES.2020.25 .9.2000173). Further studies have shown that the sensitivity of assays for the detection of SARS-CoV-2 RNA differs and can differ by more than 1 x log10 (Matheeussen V et al., Eurosurveill., Jul 9; Görzer et al., J Clin Virol 2020: 129, 104537). The detection limit depends on the target sequence used, but also on other factors. The manufacturer's information must be taken into account for the respective assay and verified by comparative measurements in the laboratory before the test is introduced. The ÖGLMKC recommends the use of an assay with a sufficiently low detection limit and a corresponding review of the entire test performance as part of external quality assurance (including samples with low viral load in round-robin test programs). The ÖGLMKC expressly recommends (if available) the use of CE/IVD-certified assays for the detection of SARS-CoV-2 RNA. If in individual cases it is necessary to introduce an in-house PCR test for the detection of SARS-CoV-2 RNA because CE-marked tests are not available, the test performance must be thoroughly validated and documented by the performing laboratory. 1.1.4. Assessment of PCR resultsThe duration of detection of viral RNA in the nasopharynx secretion seems to be subject to large individual fluctuations and is 12 days (1-24 days) in the median, according to a series of cases. In > 80% of patients, the detection is positive for at least 7 days (JAMA. 2020 Mar 3. doi: 10.1001/jama.2020.3204). In some cases, a PCR positivity for >25 days was also described. A recent work describes a detailed time history of the viral load in different sample materials. At the beginning of the symptom, the concentration of viral RNA was high, decreased rapidly in throat swabs during the course of the disease and was typically detectable there for about two weeks, while in samples of the deep respiratory tract (induced sputum) and in the stool an extended viral excretion was observed (Nature 2020 April 1. doi: 10.1038/s41586-020-2196-x (2020)).
If a PCR test is based on the amplification of two (or more) target sequences of SARS-CoV-2 and only one is positive while the other is negative, there are several causes to consider: In these cases, the PCR result shall be evaluated in combination with the raw data (early or late amplification of the target) and the molecular genetic characteristics of the assay. If a technical error can be ruled out, in the current epidemiological situation it is recommended to consider the amplification of only one PCR target as a positive test result and thus as an indication of the presence of an infection with SARS-CoV-2. This is due to the consideration that only one human pathogenic beta coronavirus is currently circulating, that SARS-CoV-2 has a genetic diversity, that "weak-positive" or "non-evaluable" results may lead to uncertainties in reporting and regulatory procedures, and that any false-negative results could undermine adherence to isolation and quarantine measures.
A one-time negative PCR result does not 100% rule out SARS-CoV-2 infection. If there are reasonable grounds for suspicion of SARS-CoV-2 infection and initial negative PCR outcome, a re-sampling and examination should be agreed between the clinician and the laboratory physician. This recommendation is supported by series of cases according to which PCR may initially be negative in epidemiologically, clinically and CT morphologically defined suspected cases (Lancet. 2020 Feb 15;395(10223):514-523. doi: 10.1016/S0140-6736(20)30154-9. Radiology. 2020 Feb 12:200343. doi: 10.1148/radiol.2020200343.). False-negative results may be based in particular on pre-analysis and are based, for example, on poor sample quality, long sample storage, improper sample transport or unfavorable timing of sampling, based on the course of the disease. Inhibition of the PCR or mutation of the virus may play a role in analytics (Clin Infect Dis. 2020 Mar 4. pii: ciaa203. doi: 10.1093/cid/ciaa203.). The former can be detected by using an internal control for nucleic acid extraction and amplification. 1.1.4.3. Consideration of the Ct-valueIn the literature, a correlation between the viral load in the test material and the cultivability of the viruses contained in the sample in cell culture is described (Eur J Clin Microbiol Infect Dis 39, 1059-1061 (2020). Doi: 10.1007 / s10096-020-03913-9) . The PCR positivity at a later point in time in the course of the reaction (usually indicated as a high Ct value) corresponds approximately to a low viral load in the sample. The recommendation of the Ministry of Health for the "release of COVID-19 cases from isolation" of July 9th, 2020, based on the recommendations of the RKI, names a Ct value of greater than 30 as the limit for a weakly positive sample. From a laboratory diagnostic point of view, a generally valid definition of such a limit value should be questioned. Most SARS-CoV-2 PCR tests are not validated for quantification of the viral load. The Ct value of a sample is decisively influenced by the pre-analysis (type of sample collection, sample storage), the RNA extraction and the PCR method. Proficiency testing therefore shows relevant differences in the Ct value of the same sample in different laboratories (Görzer et al. Journal of Virology. Doi: 10.1016 / j.jcv.2020.104537 and Matheeussen et al. Euro Surveill. Doi: 10.2807 / 1560-7917.ES. 2020.25.27.2001223). However, a good match between the Ct values of a particular test and those of the published tests is a basic requirement for this assessment. This requires specific internal and external quality controls and should therefore also be considered separately in future round robin programs. Regardless of analytical considerations, it is essential to take into account that the relationship between the viral load (or Ct value) and the ability of the viruses to be grown in cell culture, especially in samples in the late course of the disease of COVID-19 patients (sometimes several weeks after the onset of symptoms), is ascertained has been. However, there are insufficient data on the relationship between viral load at the beginning of the disease and infectivity. Correspondingly, the considerations of the RKI regarding the consideration of the Ct value relate primarily to the discharge management of patients after the end of the symptomatic phase of illness. When interpreting the Ct value, it is therefore essential to take into account several influencing factors such as the point in time in the course of the disease and the quality of the sampling as well as the type of material or the smear location, the processing and the test system used. From the point of view of the ÖGLMKC, no reliable statement on infectivity can be derived from the Ct value alone, without taking these influencing factors into account. In this context, the development of quantitative SARS-CoV-2 PCR tests, which better correspond to the desire for a quantitative assessment of the viral load, would be required. 1.1.5. Pool testingIn so-called pool testing, a defined amount of the starting material from several individual samples is combined to form a pool and then only the resulting pool sample is analyzed using PCR. If the result is negative, it is assumed that every sample contained in the pool is to be regarded as negative. If the pool is tested positive, the pool is dissolved and the samples contained are analyzed individually. The pool testing method is primarily aimed at increasing efficiency, mostly with limited test resources (Hanel et al, arXiv: 2003.09944). To minimize the risks of a potential sample mix-up, the sample pooling itself should also be largely automated, if possible. From a laboratory point of view, it must in any case be checked whether the pool testing has been checked as part of the test approval (CE / IVD) and whether the test is approved for this application. If this is not the case, the responsibility lies with the laboratory carrying out the work and a documented validation must be carried out in the laboratory. Since the increasing dilution effect with increasing pool size leads to a loss of sensitivity and therefore samples with low virus concentrations may no longer be detected, it is recommended that the method only be used for non-event-related or epidemiological investigations, such as pool testing of COVID-19 Suspected cases are not advised. The maximum pool size is limited both by the detection limit of the test procedure and by the epidemiological situation. From an epidemiological point of view, in conjunction with the currently available literature (J Med Virol. Doi: 10.1002 / jmv.25971), it is recommended not to exceed a pool size of a maximum of 10 samples per pool, depending on the underlying prevalence. If the pool size is too high, if the prevalence is high, too many pools have to be closed and the method is no longer efficient or resource-saving. However, depending on the analytical sensitivity of the test procedure, the maximum acceptable pool size can also be significantly there are less than 10 samples. 1.2. Direct virus detection using an antigen testThe antigen test is a direct virus detection which immunologically detects viral proteins in respiratory sample materials. Point-of-care systems or quick test formats are mainly used for this. In contrast to the PCR test, the antigen test does not require any special laboratory equipment and can be carried out outside of medical laboratories. The rapid availability of the results means that rapid antigen tests are suitable for decentralized testing of symptomatic persons as part of the differential diagnosis of respiratory infections. The detection limit of antigen tests is several orders of magnitude higher than that of PCR, so that a high concentration of viral particles is required in the sample in order to reliably detect a SARS-CoV-2 infection using an antigen test. For example, the WHO has formulated a minimum detection limit equivalent to 10^6 (acceptable) or better 10^4 (desirable) genome copies/ml for antigen tests (target product profiles for priority) for the detection of acute SARS-CoV-2 infection in symptomatic persons diagnostics to support response to the COVID-19 pandemic, WHO, 2020). With its significantly better analytical sensitivity (detection limit in the range from 10^1 to 10^2 genome copies/ml), the PCR is therefore still the reference method for direct virus detection. Due to the clear differences in the detection limit, alternative methods of sampling (such as throat rinsing fluid) or sample processing (such as pool testing) that have been evaluated for PCR tests cannot be transferred to antigen tests. Sampling and preanalytics must therefore be carried out in accordance with the respective manufacturer's instructions; all deviations from this must be validated separately for the respective test. In terms of clinical performance data for the use of antigen tests in situations in which PCR testing is not available or not available quickly enough, the WHO formulates a sensitivity of ≥80% and a specificity of ≥97% as acceptable or a sensitivity in symptomatic patients of ≥90% and a specificity of ≥99% as desirable (Target product profiles for priority diagnostics to support response to the COVID-19 pandemic, WHO, 2020). When assessing the performance data given by the manufacturer, it must be taken into account whether the patient population examined is representative for the planned use of the antigen test; this also applies in particular to use in asymptomatic persons. Only a few manufacturer-independent evaluations of antigen tests are currently published. However, initial publications and our own data indicate significant differences in performance between the antigen tests examined, which emphasizes the need for manufacturer-independent validation. Suitable antigen tests can be a useful addition to the PCR test capacities where an initial (pre-) decision about the possible presence of a transmission-relevant infection should be made quickly (on site, POCT) in the early phase of the infection (Antigen detection in the diagnosis of SARS-CoV-2 infection using rapid immunoassays: interim guidance, WHO, 2020; RKI). The evaluation of the results of in vitro diagnostics basically requires expertise and the inclusion of knowledge about the test indication, the quality of the sampling and the consequences of a positive or negative result (RKI). This applies in particular to the use of antigen tests in a point-of-care setting. 1.2.1. Evaluation of the results of antigen testsA negative result of an antigen test does not rule out an infection with SARS-CoV-2. In particular, if there is a low virus concentration in the sample, as is typical in the early incubation phase and in the late phase of infection, false negative results often occur, so that the clinical sensitivity of the antigen test is inferior to that of the PCR. This must be taken into account when defining areas of application and interpreting negative results. If there is a high clinical suspicion of the presence of a SARS-CoV-2 infection, a new test using PCR is indicated if the antigen test result is negative. If an antigen test is used as part of a regular test concept, this limitation can potentially be partially compensated for by a high frequency of testing. A positive result of an antigen test speaks - with the corresponding symptoms - for the presence of an infection with SARS-CoV-2. False positive results were described to a variable extent (https://www.fda.gov/medical-devices/letters-health-care-providers/potential-false-positive-results-antigen-tests-rapid-detection-sars-cov-2-letter-clinical-laboratory; https://www.medrxiv.org/content/10.1101/2020.11.12.20230292v1). False positive test results can be excluded by confirmation using PCR. Whether this is necessary in individual cases or whether the specificity of the antigen test used is sufficient depends crucially on the probability of the pre-test, so that the current epidemiological situation and the symptoms of the person tested should be taken into account when making this decision. 1.3. Indirect virus detection using an antibody testImmunological tests for serological examination detect antibodies against coronavirus SARS-CoV-2 in the blood of patients which are formed as part of the patient's immune response against SARS-CoV-2. The time course until the detection of antibodies in the context of a SARS-CoV-2 infection can vary individually and has not yet been conclusively investigated for many tests. In general, the start of seroconversion was described about 10-14 days after the onset of symptoms (Nat Med. 2020 Apr 29; doi.org/10.1038/s41591-020-0897-1). The available antibody tests differ in essential respects:
For many antibody tests on the market, there are currently insufficient data on sensitivity and specificity, which were determined in studies independent of the manufacturer. However, new papers are being published on this subject, mainly so-called �preprints�, ie pre-publications without peer reviews, many of which should appear as reviewed scientific articles in the coming months. It is crucial for the diagnostic application that these data have been collected from a group of patients or healthy persons relevant to the clinical question. For example, data on the clinical sensitivity and specificity of a test, which was collected in COVID-19 patients in the advanced course of the disease, cannot be used for the question of a COVID-19 diagnosis at the onset of symptoms. If the indication for an antibody test is made, those tests should be used which, according to the current state of the art, have the highest specificity, sensitivity and precision and whose performance data have been verified by the respective laboratory. The ÖGLMKC therefore recommends carrying out serological tests for SARS-CoV-2 only in medical laboratories and using near-patient serological rapid tests only in exceptional cases. In summary, according to the current state of knowledge, serological tests alone (without PCR) are neither suitable for diagnostic detection nor to rule out an acute infection by SARS-CoV-2. In individual cases, however, it can make sense to carry out anti-SARS-CoV-2 antibody tests in addition to PCR analyzes, especially if several serum samples are examined with a valid antibody test during the course of the disease. Much more important are antibody tests for epidemiological studies to assess the number of unreported cases of COVID-19 infections that have not been confirmed by PCR detection. The ÖGLMKC recommends to refrain from the uncritical use of antibody tests. 1.3.1. Potential uses of antibody tests against SARS-CoV-2
1.3.2. Evaluation of a positive antibody finding A positive antibody finding should always be interpreted in the context of the specificity of the test system used, expected seroprevalence and clinical information on the patient. Several SARS-CoV-2 anti-body tests are currently available, which in very large cohorts (> 1000 individuals) have high specificities with> 99% and sometimes. Reach> 99.5%.
1.3.3. Examples for the interpretation of the results of an antibody test In the interpretation of the findings of an antibody test, the limitations according to the current data situation should be explicitly mentioned. An example of the interpretation of a positive or negative anti-SARS-CoV-2 IgG test is given below. Without prejudice to this, an individual interpretation of the findings can and should of course also take place. This can also take into account the respective prevalence of COVID-19.
The positive test result is an indication of an existing or past infection with the SARS-CoV-2 virus. Rare cross-reactivities cannot be ruled out with absolute certainty. Based on the current data, no statement can currently be made on the extent and duration of immunity.
A negative test result does not reliably rule out contact with the SARS-CoV-2 virus. The formation of IgG antibodies begins relatively early with SARS-CoV-2 (10-14 days after the onset of symptoms), however, both the time at which the onset and the amount of antibodies formed can vary individually, so that about 3 weeks after the onset of symptoms the majority of COVID-19 sufferers have specific antibodies in their blood. At the same time, more and more cases are reported in the specialist literature in which the antibody formation is very moderate and therefore does not necessarily lead to a positive result in every test system.
1.4. Definition of terms for the properties of diagnostic tests 1.4.1. Analytical sensitivity and specificity The analytical sensitivity and specificity of a test describe the suitability of a test to detect a particular analyte (e.g. SARS-CoV-2 nucleic acids) in a sample. These characteristics of a laboratory test can be determined by a pure technical validation of the test. The detection limit of a method describes the lowest concentration of the analyte, which can be reliably detected by the test. A test with a low detection limit can detect low concentrations of the analyte and has a high analytical sensitivity. The analytical specificity of a test describes the ability of the test to detect only the desired analyte and not to be influenced by other substances in the sample (in addition to general interference factors, for example, other coronaviruses). 1.4.2. False negative and false positive results Differences between the actual presence of a disease and the result of a laboratory test are called false negative or false positive results. False negative people are sick patients, which the test mistakenly classifies as healthy.False positives are actually healthy individuals, which the test mistakenly classifies as sick. Really negative people are healthy people, which the test correctly classifies as healthy. Really positive people are sick patients, which the test correctly classifies as sick. 1.4.3. Clinical sensitivity and specificity The clinical sensitivity and specificity of a test describe the suitability of a laboratory test to distinguish the sick and the healthy. These characteristics of a laboratory test can only be collected by clinical validation of the test with patient samples and are only valid for the clinical situation for which the patient group used is representative. The diagnostic sensitivity describes the proportion of correctly positive test results in patients with a disease and is usually expressed in %. A test has a high diagnostic sensitivity when few false negative results occur. The diagnostic specificity describes the proportion of correctly negative test results in healthy people and is usually expressed in %. A test has a high diagnostic specificity when few false positives results occur. 1.4.4. Positive and negative predictive value (predictive value) In addition to its diagnostic sensitivity and specificity, the predictive value of a laboratory test also depends on the frequency or prevalence of the disease in the population. As a result, the predictive value of the same laboratory test changes when the frequency of a disease such as COVID-19 increases significantly in the population. The positive predictive value is the probability of a disease if the laboratory test is positive (pathological). The higher the clinical specificity of a laboratory test and the more frequent a disease occurs, the higher the positive predictive value of the test. The negative predictive value is the probability that a disease can be ruled out if the laboratory test is negative (normal). The higher the clinical sensitivity of a laboratory test and the less frequently a disease occurs, the higher the negative predictive value of the test. The testing of sample material for human diagnostic purposes with test kits and devices is subject to the Austrian Medical Devices Act (MPG) and its aim is to ensure the safety and high-quality care of patients and society with medical devices and laboratory diagnostic tests. Compliant, high-quality and therefore legally permissible testing therefore requires
The requirements of the MPG, including quality assurance, apply to every laboratory that carries out tests for patients, regardless of the legal status or any crisis situations, and are not overridden by the Epidemic Act or related regulations. The Austrian Medical Devices Act is based on the EU Directives 93/42 EG and 98/79 EG, within the framework of which national law must comply. Quality assurance in medical laboratories in the established area is determined by an ordinance of the Medical Association (approved by the Ministry of Health). Quality assurance in medical laboratory analysis in the hospital sector is included in the Cure and Hospital Act. The ÖNORM K 1950 gives a qualified recommendation for the concrete implementation based on the international standard EN ISO 15189: 2014. The relevant regulations on documentation for the traceability and liability of the medical laboratories are contained in part in the Medical Devices Act, in part in the Physicians Act and in the Health and Medical Institutions Act and are important for patients and authorities in order to be able to make inquiries and, if necessary, assert claims . The structural and quality requirements laid down in the regulations are not only legally required for the correct medical high-quality performance of laboratories, but also one of the most important requirements for the work of clinically active doctors, who rely on medically meaningful findings and accompanying expertise for their work are instructed on the patient. The Agency for Health and Food Security (AGES) is legally obliged (�68 MPG) to monitor all laboratories that use medical devices (in-vitro diagnostics) to ensure compliance with these legal provisions and to act accordingly in the event of violations to ensure that patients are at risk and can be prevented from the public by faulty laboratory analysis. 1.5.2. Assessment of diagnostic test In the current pandemic situation, the test kits available on the market (in vitro diagnostics) have either been approved with urgent approvals for the American market (FDA) and then CE-marked by the manufacturers, or are only available as research kits for which there is no comprehensible verification the quality has been achieved by the manufacturer, whereby manufacturers can now only be monitored very incompletely, especially with SARS-CoV-2 kits, and are actually de facto autonomous when applying the CE label. This means increased caution is also required when carrying out CE-marked tests. CE-marked tests for SARS-CoV-2 diagnostics in the laboratory are, according to the current legal situation, not subject to the need for an objective evaluation of the manufacturer's information by an independent notified body. For research kits, each laboratory must assume full responsibility for the analysis and clinical usability of the findings (in-house test), i.e. carry out the validation / evaluation itself, including acceptance and transport of the samples intended for the test, which are otherwise carried out by the industrial manufacturers in complex test procedures. The laboratory must document that the performance requirements in Annex I of EU Directive 98/79 EC have been met and that the documentation can be submitted to the authority if necessary. The European Commission has summarized the current status of the literature on performance data from SARS-CoV-2 Test: "Current performance of COVID-19 test methods and devices and proposed performance criteria - Working document of Commission services" (Link: https://ec.europa.eu/docsroom/documents/40805). This document also emphasizes that for many of the laboratory tests there are no independent studies to assess the performance data. 1.5.3. Validation and verification of laboratory tests Every laboratory test that is to be used for diagnostic purposes on human samples must be checked in advance by the laboratory for its suitability. This also applies to CE-marked tests. The EN ISO 15189: 2014 standard differentiates between validation and verification of the test procedure.
In the case of CE-marked laboratory tests for SARS-CoV-2, the manufacturer is obliged to provide the performance data of the test. The quality of this information varies widely among the tests currently available and is currently not being verified by an independent notified body. It is therefore the responsibility of the laboratory to check the quality of the performance data specified by the manufacturer and to assess whether the validation was carried out by the manufacturer according to scientific standards. In addition, the achievement of the performance data must be verified in the respective laboratory (see validation and verification of laboratory test). In practice, there are common problems with manufacturer information, which will be dealt with as examples. 1.5.5. Selection of subjects to ascertain clinical sensitivity and specificity Ideally, clinical sensitivity and specificity of an examination procedure should be ascertained on a patient who has been coughed up, which is representative of the clinical question and also allows an assessment of the positive or negative predictive value of the test. In practice, due to the limitations of the test-independent definition of COVID-19 patients, almost all manufacturers use two independent patient cohorts to ascertain the clinical sensitivity and specificity. For example, the clinical sensitivity of an anti-SARS-CoV-2 antibody test in patients with PCR-related COVID-19 disease is assessed. Irrespective of this, sera from test persons who had been archived before the occurrence of SARS-CoV-2 are used to determine the specificity. This procedure makes sense in the current situation, but has certain limitations; in particular, no direct conclusions can be drawn from such studies that include the prevalence of SARS-CoV-2. It is expressly forbidden to use the sum of these two cohorts to calculate a positive or negative predictive value of the test, as is done by individual manufacturers. Such an approach would equate the prevalence of COVID-19 with that resulting from any combination of these two cohorts.
1.5.6. Insufficient number of cases and missing information about confidence intervals "Current performance of COVID-19 test methods and devices and proposed performance criteria - Working document of Commission services" (Link: https://ec.europa.eu/docsroom/documents/40805), emphasizes the need for 95% confidence intervals for the Specify the results of diagnostic sensitivity and specificity of a laboratory test, which was examined in a clinical study using a suitable cohort. In this context, the number of cases of the examined subjects is significant. Figure 1 shows the relationship between the number of cases and the width of the confidence interval in a test with 95% specificity according to Bruderer (https://onlinelibrary.wiley.com/doi/epdf/10.1111/j.1553-2712.1996.tb03538.x). 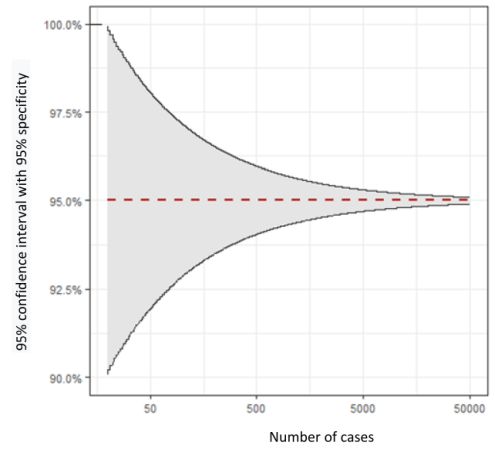
Figure 1:Example of the relationship between the width of the 95% confidence interval and the number of cases, with the following assumptions: 95% specificity, error probability α=0.05, 1% prevalence For example, if a manufacturer checks the diagnostic specificity of an Anti-SARS-CoV-2 antibody test using only 50 subjects and no false positive result is shown for these 50 samples, the validity of this test evaluation is relatively low, although the specificity is nominally 100% . Individual manufacturers have evaluated anti-SARS-CoV-2 antibody tests in more than 1,000 subjects, which leads to more statistically meaningful results with a narrow confidence interval. 1.5.7. External quality controls Round robin tests are an essential means of external quality assurance. For this purpose, samples are sent from an external point to the participating laboratories. The interlaboratory test samples are to be used and processed in the laboratory like patient samples. The results are reported back by the laboratory and assessed by the proficiency testing manager. Both SARS-CoV-2 PCR tests and anti-SARS-CoV-2 antibody tests are now available in round robin tests from different providers. The interlaboratory test samples are to be used and processed like patient samples. The results are assessed by the proficiency testing management. The ÖGLMKC advocates mandatory participation in proficiency testing for SARS-CoV-2 tests. For all laboratories that carry out SARS-CoV-2 analyzes on humans without exception, sanctionable regulations to ensure the required quality in the use of in-vitro diagnostics are essential. 1.5.7.1. Assessment of results in SARS-CoV-2 PCR round robin tests Published data from the INSTAND round-robin test SARS-CoV-2 virus genome detection April 2020 have led to speculations about the specificity of the SARS-CoV-2 PCR. In this round-robin test, 1.4% false positive results were reported for negative samples. However, the data from a round-robin test cannot be applied directly to the specificity data of an assay; rather, probes reflect the quality of the participating laboratories. This 1.4% relates to a few laboratories that have reported incorrect results. According to the assessment of the ÖGLMKC and the ÖQUASTA False negative results are frequently observed in samples with low virus levels in round trials (Görzer et al. Journal of Virology. Doi: 10.1016 / j.jcv.2020.104537). This may indicate that the test used is not sensitive enough to reliably identify samples with a low viral load as positive. For such laboratories and tests, the ÖGLMKC assesses the use of pool testing as critical. Such interlaboratory test results clearly show how important the quality of the testing laboratories is. Against this background, the ÖGLMKC is critical of the current legal situation that allows non-medical laboratories to carry out SARS-CoV-2 tests in the context of the COVID-19 pandemic on the basis of the Epidemic Act. Trained specialists, extensive experience with molecular infection diagnostics, effective systems of quality assurance and medical reports are basic requirements to ensure high quality care in Austria. The highest medical quality standards must be uncompromisingly demanded by laboratories that carry out SARS-CoV-2 PCR tests. 2. General laboratory diagnostics for COVID-19 2.1. Handling of blood samples and other body fluids from COVID-19 patients In addition to respiratory secretions, coronavirus SARS-CoV-2 was also detected in the stool and in isolated cases in urine, blood/plasma and cerebrospinal fluid by means of PCR. Although no contagion via these bodily fluids (urine, blood/plasma and cerebrospinal fluid) is documented, such a must not be ruled out with the last certainty at present. Based on the current data situation, ÖGLMKC therefore recommends that general hygiene and protective measures be taken into account when working with blood samples from COVID-19 patients and that unnecessary aerosol formation during sample handling should be avoided. For stool samples of COVID-19 patients, the potential risk of infection is currently assessed higher, so the ÖGLMKC recommends reducing the analysis of stool samples from COVID-19 patients or suspected cases to an absolutely necessary minimum and taking additional protective measures to minimize the risk of potential infection. In the course of a laboratory request, the laboratory must in principle be notified by the sender that the submission is samples of a COVID-19 patient or a COVID-19 suspected case. In detail, we allow ourselves to refer to the recommendations for handling test material from Covid 19 positive / suspect patients in the laboratory of the Austrian Society for Hygiene, Microbiology and Preventive Medicine (ÖGHMP): 2.2. Value of laboratory parameters with COVID-19 Clinical and laboratory data showed that some laboratory values are frequently changed in COVID-19 (see summary in Lippi G, Plebani M. Clin Chim Acta. 2020 Mar 4. https://doi.org/10.1016/j.cca.2020.03.004). Among the hematological parameters, it was found that lymphopenia in particular is associated with the severity of the course of the disease. Patients suffering from COVID-19 with lymphocytopenia had a poor prognostic course (Tan L et al. Signal Transduct Target Ther. 2020 Mar 27; 5:33. https://doi:10.1038/s41392-020-0148-4) . In most cases, there was a reduction in CD8 positive cytotoxic T suppressor cells (Liu Y et al. Sci China Life Sci. 2020 Mar; 63: 364�374. https://doi.org/10.1007/s11427-020-1643-8; Velavan TP et al. Int J Infect Dis 2020 Jun; 95: 304-307. https://doi:10.1016/j.ijid.2020.04.061). Patients with severe disease had more frequent liver dysfunction with increased alanine (ALT) and aspartate aminotransferases (AST) levels compared to mild forms (Zhang C et al. Lancet Gastroenterol Hepatol. 2020 May; 5: 428-430. https://doi:10.1016/S2468-1253(20)30057-1). The severity and prognosis of the course of the disease showed a clear association with increased inflammatory markers, in particular the C-reactive protein (CRP), procalcitonin and ferritin (Terpos E. Am J Hematol. 2020 Jul; 95: 834-847. https://doi:10.1002/ajh.25829). In a current study, a D-dimer = 2.0 µg / mL (4-fold increase) at the time of hospitalization proved to be a predictive mortality marker (Zhang L. J Thromb Haemost. 2020 Jun; 18: 1324-1329. https://doi:10.1111/jth.14859). Compared to healthy people, COVID-19 sufferers showed significantly increased D-dimer and fibrinogen levels as well as greatly reduced antithrombin levels as a sign of impaired blood clotting (Han H. Clin Chem Lab Med. 2020 Jun 25; 58: 1116-1120. https://doi:10.1515/cclm-2020-0188). An elevated troponin value turned out to be a predictive marker for mortality in patients with COVID-19 pneumonia (Du RH. Eur Respir J. 2020 May 7; 55: 2000524. https://doi:10.1183/13993003.00524-2020). Some studies have also examined the importance of cytokine determination in the treatment of COVID-19 patients. In particular, IL-6 and TNF-alpha were identified as independent and prognostically relevant biomarkers for patients. The study authors recommend that these cytokine determinations should be taken into account for the management of patients and the guidance of therapies (DM Del Valle, PMID: 32511562). With regard to the value of laboratory diagnostic parameters in clinical decision-making, further large data sets and meta-analyzes are necessary. The ÖGLMKC recommends a structured collection and evaluation of laboratory data from Austrian COVID-19 cases, taking into account previous publications. This is intended to bundle the experiences of the various health facilities and to subject the prognostic significance of the individual biomarkers to a further evaluation. Federal Ministry of Social Affairs, Health, Care and Consumer Protectionhttps://www.sozialministerium.at/Informationen-zum-Coronavirus/Neuartiges-Coronavirus-(2019-nCov).html Agency for Health and Food Safety (AGES) https://www.ages.at/themen/krankheitserreger/coronavirus/ Robert Koch Institute (RKI) https://www.rki.de/DE/Content/InfAZ/N/Neuartiges_Coronavirus/nCoV.html?cms_box=1&cms_current=COVID-19+%28Coronavirus+SARS-CoV-2%29&cms_lv2=13490882 World Health Organization (WHO) https://www.who.int/emergencies/diseases/novel-coronavirus-2019 Austrian Society for Hygiene, Microbiology and Preventive Medicine (ÖGHMP) https://www.oeghmp.at/ 4. Contact details of laboratories for the detection of SARS-CoV-2 in Austria The ÖGLMKC has launched a website of specialist medical laboratories for SARS-CoV-2 diagnostics, which lists detailed information on the methodology used and contact data of the laboratories and offers a search and filter function. Note for laboratories: Interested specialist laboratories who agree to the publication of their contact details on the website mentioned above, please contact the ÖGLMKC office. You can find more information on the website. https://www.covid19-labore.at/ All laboratories that carry out SARS-CoV-2 diagnostics are urged to take part in round trials as part of external quality assurance. In Austria, the Austrian Society for Quality Assurance and Standardization of Medical Diagnostic Examinations (ÖQUASTA
Central Institute for Medical and Chemical Laboratory Diagnostics, University Hospital Innsbruck & Clinical Institute for Laboratory Medicine, Medical University of Vienna Postal address: Universitätskliniken Innsbruck, ZIMCL, Anichstraße 35, 6020 Innsbruck Tel: +43 (0) 512 504 - 83673 E-Mail: gregor.hoermann@tirol-kliniken.at; gregor.hoermann@meduniwien.ac.at | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||